Universal first step: EstimateFractions()
Source:vignettes/EstimateFractions.Rmd
EstimateFractions.RmdIntroduction
The first step of almost any NR-seq analysis is estimating how much
of each mutational population is present for each feature in each
sample. For example, in a standard TimeLapse-seq/SLAM-seq/TUC-seq/etc.
experiment, you need to first estimate the fraction of reads from each
feature that have high T-to-C mutational content.
EstimateFractions() is designed for exactly this task. In
this vignette, I will walk through the basics of using
EstimateFractions(), while also diving into some of its
unique functionality.
library(EZbakR)
# Going to show one tidyr trick to help with cB filtering
library(tidyr)
# Going to use a bit of dplyr magic in my demonstrations
library(dplyr)
#>
#> Attaching package: 'dplyr'
#> The following objects are masked from 'package:stats':
#>
#> filter, lag
#> The following objects are masked from 'package:base':
#>
#> intersect, setdiff, setequal, unionThe basics
First, let’s simulate some data to showcase how
EstimateFractions() works:
simdata <- EZSimulate(nfeatures = 300, nreps = 2)
ezbdo <- EZbakRData(simdata$cB, simdata$metadf)Technically, all you need to do is run
EstimateFractions(), providing it your
EZbakRData object, and all will be fine:
ezbdo <- EstimateFractions(ezbdo,
features = "feature")
#> Estimating mutation rates
#> Summarizing data for feature(s) of interest
#> Averaging out the nucleotide counts for improved efficiency
#> Estimating fractions
#> Processing output
# NOTE:
# For real data from fastq2EZbakR, you will often
# want to set `features = "XF"`, specifying analysis
# of reads mapping to exonic regions of genes.Despite this, it is important to realize that
EstimateFractions() is automating a lot of things under the
hood. In particular, it is making the following assumptions about your
data:
- You want to analyze every mutational population tracked in your cB file. In this case, that is just the “TC” column of the simulated cB.
- All possible mutational populations are present in your data.
- The feature sets for which you want to estimate fractions includes each unique combination of all feature columns in your cB. In this case, that is just the “feature” column of the simulated cB.
- You want to filter out rows for which all feature columns are the string “__no_feature” or “NA”.
- Rows with multiple feature assignments in a given feature column should not be split into separate rows for each feature assignment.
In the rest of this section, I will discuss how to adjust all of these behaviors.
Tuning the mutational modeling
If you have multiple mutation-type columns in your cB, but would only
like to analyze a subset of them, this subset can be specified using the
mutrate_populations argument:
ezbdo <- EstimateFractions(ezbdo, mutrate_populations = "TC")
#> Estimating mutation rates
#> Summarizing data for feature(s) of interest
#> Averaging out the nucleotide counts for improved efficiency
#> Estimating fractions
#> Processing outputIf you check ?EstimateFractions(), you will see that its
default value is “all”, which means to use all present mutation-type
columns.
In addition, under the hood, EstimateFractions() creates
what I have termed a fractions design matrix. This “design matrix” is
really just a data frame with n + 1 columns, where n is the number of
mutation-types you are analyzing. EZbakR provides some lazily loaded
examples for you to check out.
“Standard” TC-only fractions design matrix:
# Observe contents of cB
standard_fraction_design| TC | present |
|---|---|
| TRUE | TRUE |
| FALSE | TRUE |
There is one column for each mutation-type, and one column called
present. Each value under a mutation-type column is either
TRUE or FALSE. TRUE indicates
that the row describes a population with high levels of that
mutation-type. So the first row in this example describes the high
T-to-C mutation content population. The value under the
present column denotes whether or not that population is
expected to be present in your data. The first row has a
present value of TRUE because we expect there
to be high T-to-C mutation content reads in our +label data (don’t worry
about -label controls, they are properly handled automatically). The
second row has a TC value of FALSE, indicating
that it pertains to the population of reads with low T-to-C mutation
content. This population is also expected to exist in a standard NR-seq
dataset, so its present value is also `TRUE.
For a more complicated example, consider the fractions design matirx for TILAC. TILAC is a method where an s4U labeled RNA population is mixed with an s6G labeled population. In this case, there are expected to be reads with either high T-to-C content (new reads from the s4U labeled sample), high G-to-A content (new reads from the s6G labeled sample), and reads with low T-to-C and G-to-A mutational content (old reads from either sample). However, you will NEVER expect a read with both high T-to-C and high G-to-A content, as there are no samples subjected to both labels:
# Observe contents of cB
tilac_fraction_design| TC | GA | present |
|---|---|---|
| TRUE | TRUE | FALSE |
| TRUE | FALSE | TRUE |
| FALSE | TRUE | TRUE |
| FALSE | FALSE | TRUE |
Because of this the present value for the
TC == TRUE and GA == TRUE row is
FALSE. The dually high mutation population is not present
in this data. All other populations are present though, so their
present values are TRUE.
You can automatically generate a fraction design table as such:
# Three populations for fun:
fd_table <- create_fraction_design(c("TC", "GA", "CG"))
fd_table| TC | GA | CG | present |
|---|---|---|---|
| TRUE | TRUE | TRUE | TRUE |
| FALSE | TRUE | TRUE | TRUE |
| TRUE | FALSE | TRUE | TRUE |
| FALSE | FALSE | TRUE | TRUE |
| TRUE | TRUE | FALSE | TRUE |
| FALSE | TRUE | FALSE | TRUE |
| TRUE | FALSE | FALSE | TRUE |
| FALSE | FALSE | FALSE | TRUE |
By default, create_fraction_design assumes all possible
populations are present. You can edit this table to better reflect the
true circumstances of your particular experiment. If you don’t provide a
fraction design table, EstimateFractions() will use the
default output of create_fraction_design() to ensure that
all possible populations are modeled. This is rarely going to be a truly
accurate fraction design table for multi-label experiments, but its a
conservative default that you can easily adjust.
Tuning the feature set choice and filtering
fastq2EZbakR, the upstream pipeline I developed to conveniently
produce output compatible with EZbakR, is able to assign reads to lots
of different “features”. These include genes, exonic regions of genes,
transcript equivalence classes, exon-exon junctions, and more. To make
full effective use of this diverse set of feature assignments, its
important to understand how EstimateFractions() will treat
the various feature columns in your cB. Often, you will be interested in
analyzing subsets of these features separately. For example, it might
make sense to estimate fractions for gene + transcript equivalence class
combos, gene + exon-exon junction combos, and the standard exonic-gene
feature, all with the same cB. You can specify which features to use in
a given analysis via setting the features argument:
ezbdo <- EstimateFractions(ezbdo, features = 'feature')
#> Estimating mutation rates
#> Summarizing data for feature(s) of interest
#> Averaging out the nucleotide counts for improved efficiency
#> Estimating fractions
#> Processing outputIn this example, there is only one feature column called
feature, but in practice, the features
argument can be provided a vector of feature column names.
Sometimes, a read was not assignable to a given feature. In that
case, there is likely a characteristic string that denotes failed
assignment. In the current version of fastq2EZbakR, this is
"__no_feature" (though in older versions was
NA, which is a bit harder to deal with; more on that
later). The default is thus for EstimateFractions() to
filter out any rows of the cB for which all analyzed features have this
value. This is set by the remove_features argument, which
is a vector of strings that should be considered ripe for filtering:
ezbdo <- EstimateFractions(ezbdo, remove_features = c("__no_feature", "feature1"))
#> Estimating mutation rates
#> Summarizing data for feature(s) of interest
#> Averaging out the nucleotide counts for improved efficiency
#> Estimating fractions
#> Processing outputYou can also use this to filter out certain features, like “feature1”
in the simulated example. As I mentioned though, all analyzed feature
columns need to have one of the remove_features strings in
order for them to get filtered. This behavior can be changed to the
opposite extreme, where only a single feature needs to fail this test
for the entire row to get filtered out:
ezbdo <- EstimateFractions(ezbdo, filter_condition = `|`)
#> Estimating mutation rates
#> Summarizing data for feature(s) of interest
#> Averaging out the nucleotide counts for improved efficiency
#> Estimating fractions
#> Processing outputAs I mentioned, older versions of fastq2EZbakR and bam2bakR would
denote failed assignment with an NA. While the default
value for remove_features includes the string
"NA", this does not properly handle filtering out of
columns with the actual value NA. You can convert NA’s in
your cB file with tidyr::replace_na() to whatever string
you please:
example_df <- data.frame(feature = c('A', NA, 'C'),
other_feature = c(NA, 'Y', 'Z'))
replaced_df <- replace_na(example_df,
list(feature = '__no_feature',
other_feature = 'NA'))
replaced_df| feature | other_feature |
|---|---|
| A | NA |
| __no_feature | Y |
| C | Z |
Finally, some feature assignments will be ambiguous. That is, one
read will assign to multiple instances of a given feature. For example,
when assigning reads to exon-exon junctions, one read may overlap
multiple exon-exon junctions. In fastq2EZbakR, such instances are
handled by including the names of all features a read assigned to,
separated by “+”. In some cases, you will want to split these rows into
multiple rows for each feature assignment. To do that, you can specify
the split_multi_features and
multi_feature_cols arguments:
ezbdo <- EstimateFractions(ezbdo,
split_multi_features = TRUE,
multi_feature_cols = "feature")
#> Estimating mutation rates
#> Summarizing data for feature(s) of interest
#> Averaging out the nucleotide counts for improved efficiency
#> Estimating fractions
#> Processing outputsplit_multi_features = TRUE will copy the data for
multi-feature assignment rows for any of the feature columns denoted in
multi_feature_cols. If the feature names for multi-feature
assigned reads is different from “+”, this can be addressed by altering
the multi_feature_sep argument.
Improving estimates
Two things can impact the accuracy of fractions estimation:
- Dropout: this is the phenomenon by which metabolically labeled RNA or sequencing reads derived from such RNA are lost either during library preparation or during computational processing of the raw sequencing data. Several labs have independently identified and discussed the prevalence of dropout in NR-seq and other metabolic labeling RNA-seq experiments. Recent work suggests that there are three potential causes for this: 1) Loss of labeled RNA on plastic surfaces during RNA extraction, 2) RT falloff due to modifications of the metabolic labeling made by some NR-seq chemistries, and 3) loss of reads with many NR-induced mutations due to poor read alignment.
- Inaccurate mutation rate estimates
In this section, we will discuss some strategies you can use to address these two challenges.
Correcting for dropout
If you have -label data to compare to, EZbakR can use this data to quantify and correct for dropout. Currently, the default strategy used by EZbakR is similar to that implemented in grandR and originally described here. Alternatively, EZbakR also implements that previously implemented in bakR.
Internal benchmarks suggest that that grandR strategy is a bit conservative (overestimates dropout) but more robust than the bakR strategy. The bakR strategy has the advantage of being explicitly derived from a particular generative model of dropout (details here), allowing for things like model fit assessment. This sort of functionality is currently not implemented in EZbakR though, so I have chosen to default to the grandR strategy for now.
To implement dropout correction, run CorrectDropout(),
specifying which metadf sample characteristic columns should be used to
associate -label and +label samples:
ezbdo <- CorrectDropout(ezbdo,
grouping_factors = "treatment")
#> Estimated rates of dropout are:
#> sample pdo
#> 1 sample1 0.01000000
#> 2 sample2 0.01000000
#> 3 sample3 0.01000000
#> 4 sample4 0.01101129grouping_factors is technically not a required argument,
as EZbakR will try to infer it automatically. EZbakR assumes that all
metadf sample characteristic columns should be used for grouping though,
so its important to know that this default can be altered as necessary.
To alter the correction strategy, set the strategy
parameter of CorrectDropout() (options being “grandR” and
“bakR”).
Inferring background mutation rates from -label data
In some cases (e.g., if label incorporation rates are low, or if a
very short label time was used), it can be difficult to estimate the
high and low mutation rates in a given sample. To curtail this
challenge, you can use -label data to infer background mutation rates,
and then estimate the chemically induced mutation rate fixing the
background mutation rate to the -label rate. To do this, you just need
to set pold_from_nolabel to TRUE:
ezbdo <- EstimateFractions(ezbdo,
pold_from_nolabel = TRUE)
#> Estimating mutation rates
#> Summarizing data for feature(s) of interest
#> Averaging out the nucleotide counts for improved efficiency
#> Estimating fractions
#> Processing outputBy default, a single background mutation rate (pold) will be
estimated using all of the -label data. Alternatively, you can specify
sample characteristics by which to stratify -label data, so that one
pold is estimated per group of samples with the same value for the
metadf columns specified in grouping_factors:
ezbdo <- EstimateFractions(ezbdo,
pold_from_nolabel = TRUE,
grouping_factors = 'treatment')
#> Estimating mutation rates
#> Summarizing data for feature(s) of interest
#> Averaging out the nucleotide counts for improved efficiency
#> Estimating fractions
#> Processing outputFancy functionalities
In the first section, we discussed the basic functionality of
EstimateFractions() and how to alter its key behaviors. In
this section, I will discuss some of the cooler, more niche analysis
strategies that EstimateFractions() can implement.
Hierarchical mutation rate model
The two-component mixture modeling strategy implemented in tools like bakR and GRAND-SLAM, is largely considered the “gold-standard” in analyzing NR-seq data. Like any statistical method though, it makes assumptions, and real data will often violate these assumptions. Namely, these models typically assume that every RNA synthesized in the presence of metabolic label has an equal probability of incorporating said label. What if this is not true though?This may seem like a pedantic statistical question, but there is evidence that some RNA’s deviate strongly from this assumption. For example, it has been noted (paper from the Churchman lab here) that mitochondrial RNA have much lower mutation rates in reads from new RNA than other RNA.
To account for this heterogeneity, EstimateFractions()
can implement what I will term a “hierarchical” two-component mixture
model. “Hierarchical” means that a new read mutation rate will be
estimated for each feature, but this estimate will be strongly informed
by the sample-wide average new read mutation rate. In other words, if a
given feature has enough coverage and strong evidence for its new read
mutation rate being different from the sample-wide average, then this
feature-specific mutation rate will be estimated and used in estimating
the fraction of high mutation content reads for this feature. If a
feature has limited coverage though, its new read mutation rate estimate
will be strongly pushed towards to the sample-wide average. In other
words, the feature-specific mutation rates are “regularized” towards the
sample-wide average. This strategy will take a bit longer to run and can
be implemented as so:
ezbdo <- EstimateFractions(ezbdo,
strategy = 'hierarchical')
#> Estimating mutation rates
#> Summarizing data for feature(s) of interest
#> Averaging out the nucleotide counts for improved efficiency
#> Estimating fractions
#> FITTING HIERARCHICAL TWO-COMPONENT MIXTURE MODEL:
#> Estimating distribution of feature-specific pnews
#> Estimating fractions with feature-specific pnews
#> Processing outputWhen doing this, the feature-specific and sample-wide mutation rates
will both be saved in the ezbdo$mutation_rates list.As
usual, let’s check the results:
est <- EZget(ezbdo, type = 'fractions')
truth <- simdata$PerRepTruth
compare <- dplyr::inner_join(est, truth, by = c('sample', 'feature'))
plot(compare$true_fraction_highTC,
compare$fraction_highTC)
abline(0,1)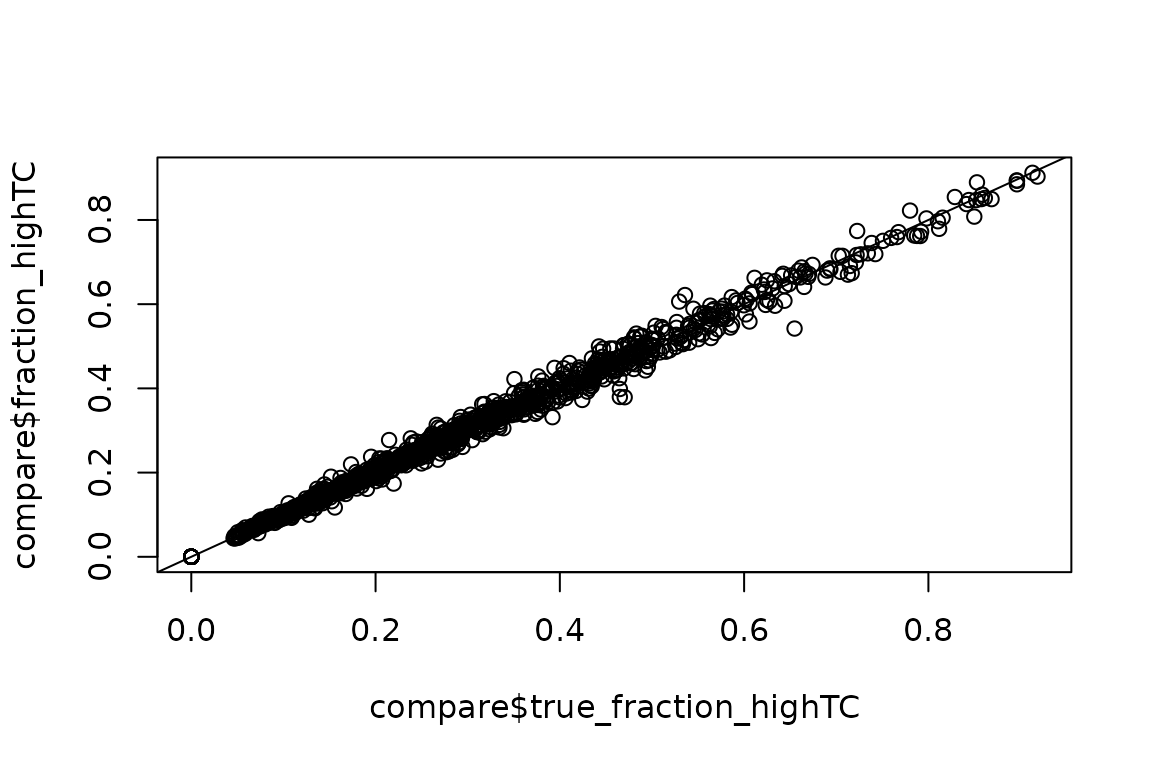
Still good!
Isoform deconvolution
For many years now, we have had tools to estimate transcript isoform abundances from RNA-seq data. These include RSEM, kallisto, Salmon, and many more. Wouldn’t it be nice if we could similarly estimate synthesis and degradation rate constants for individual transcript isoforms with NR-seq data? EZbakR now implements such an analysis strategy! To use it, you will need to have a cB in which reads have been assigned to their transcript equivalence class, which is just a fancy way of saying “the set of isoforms with which they are completely consistent”. fastq2EZbakR is able to do this, so check out its documentation for details. We can simulate such a cB ourselves for demonstration purposes:
# Simulates a single sample worth of data
simdata_iso <- SimulateIsoforms(nfeatures = 300)
# We have to manually create the metadf in this case
metadf <- data.frame(sample = 'sampleA',
tl = 4,
condition = 'A')
ezbdo <- EZbakRData(simdata_iso$cB,
metadf)If you inspect the cB simulated by SimulateIsoforms(),
you will note that there is a column called transcripts,
which represents transcript equivalence classes. The transcript IDs for
all of the transcripts a read is consistent with are separated by “+”’s,
though in this case, we don’t want to separate and copy the data for
each isoform. Whether, we want to estimate fractions for each unique
equivalence class:
ezbdo <- EstimateFractions(ezbdo)
#> Estimating mutation rates
#> Summarizing data for feature(s) of interest
#> Averaging out the nucleotide counts for improved efficiency
#> Estimating fractions
#> Processing outputTo get isoform-specific estimates, we will need to run a new
function, called EstimateIsoformFractions(). Before doing
this though, we need to import transcript isoform quantification
estimates from tools like RSEM, which will be used by
EstimateIsoformFractions(). To do this, you can use
ImportIsoformQuant(). ImportIsoformQuant() has
three parameters:
-
obj: TheEZbakRDataobject you want to which you want to add isoform quantification information -
files: A named vector of paths to each isoform quantification output file you want to import. The names should be the relevant sample names as they appear in your cB. -
quant_tool: The tool you used for isoform quantification.
ImportIsoformQuant() is just a convenient wrapper for
tximport::tximport, so I urge you to read that tools’s
documentation for more information (documentation here).
In this example, I will hack a solution, since I don’t have any
isoform quantification output to use. You could technically do something
similar if you are having a tough time using
ImportIsoformQuant(), but I wouldn’t recommend it:
### Hack in the true, simulated isoform levels
reads <- simdata_iso$ground_truth %>%
dplyr::select(transcript_id, true_count, true_TPM) %>%
dplyr::mutate(sample = 'sampleA',
effective_length = 10000) %>%
dplyr::rename(expected_count = true_count,
TPM = true_TPM)
# Name of table needs to have "isoform_quant" in it
ezbdo[['readcounts']][['simulated_isoform_quant']] <- reads
### Perform deconvolution
ezbdo <- EstimateIsoformFractions(ezbdo)
#> Analyzing sample sampleA...We can then see how it did by comparing to ground truth:
est <- EZget(ezbdo,
type = 'fractions',
features = "transcript_id")
truth <- simdata_iso$ground_truth
compare <- truth %>%
dplyr::inner_join(est, by = c("feature", "transcript_id"))
plot(compare$true_fn,
compare$fraction_highTC)
abline(0,1)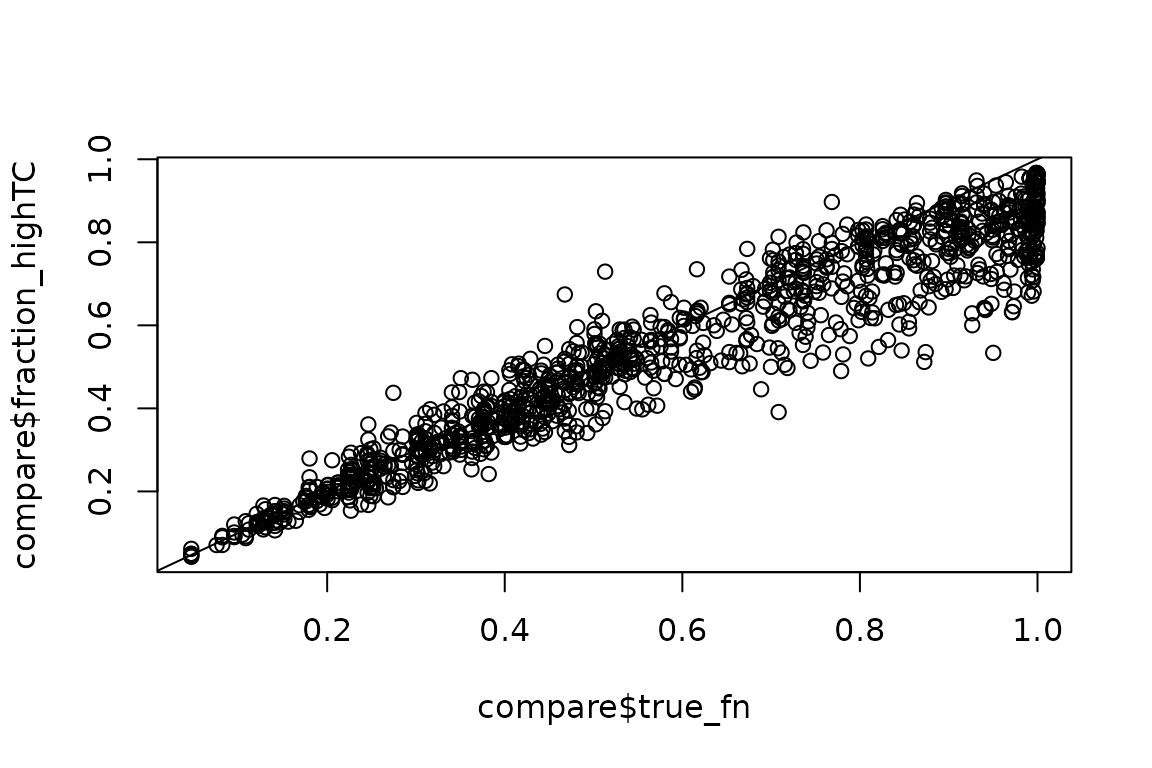
This simulation is a bit extreme as around 50% of all isoform abundance differences are assumed to be completely driven by differences in isoform stability, which leads to underestimation of some of the crazy extreme fraction news. Other than that though, it looks good!
Using the Apache Arrow backend
NOTE: The documentation for this section lacks good examples as I don’t yet have an example external dataset included with EZbakR’s installation. Thus, I will be describing the details of how the analysis would go without showing a real analysis. Look for this to change in the not-to-distant future.
What if you have a massive dataset with 10s of samples that you would like to analyze? Loading the entire cB table for such a dataset into memory will surely crash most laptops. If you have access to some sort of HPC cluster, you could of course use that, but there is nothing like the convenience and interactivity of working with a dataset on your personal computer. Because of this, EZbakR is able to use Apache Arrow’s R frontend (i.e., the arrow package) to help with large datasets. The steps for this process are described below.
Step 1: Create an Arrow Dataset
You need to create an Arrow Dataset partioned by the “sample” column
of your cB. This will create a set of .parquet files, with one file
created for each sample. This will allow
EstimateFractions() to load your data in one sample at a
time, so as to only hold a single sample of the cB in RAM at a time:
library(arrow)
### Move into the directory with your cB file
setwd("Path/to/cB/containing/directory")
### This will not load the cB into memory
# You can run `read_csv("cB.csv.gz", n_max = 1)` to check to see what
# the order of the columns are, as this order needs to match your provided
# schema.
ds <- open_dataset("cB.csv.gz",
format = "csv",
schema = schema(
sample = string(),
rname = string(),
GF = string(),
XF = string(),
exon_bin = string(),
bamfile_transcripts = string(),
junction_start = string(),
junction_end = string(),
TC = int64(),
nT = int32(),
sj = bool(),
n = int64()
),
skip_rows=1)
### Create Arrow dataset
setwd("Path/to/where/you/want/to/write/Arrow/Dataset")
ds %>%
group_by(sample) %>%
write_dataset("fulldataset/",
format = "parquet")See the arrow documentation for a lot more details about how to tune the dataset creation process. This will never load the entire cB into memory, though may use a bit too much RAM if you try to add some of the custom filtering or summarization discussed in the arrows docs.
Step 2: Create EZbakRArrowData object
You can then create an EZbakRArrowData object similarly
to how you would create a standard EZbakRData object:
ds <- arrow::open_dataset("Path/to/where/you/want/to/Arrow/Dataset/")
metadf <- tibble(sample = c("WT_1", "WT_2", "WT_ctl",
"KO_1", "KO_2", "KO_ctl"),
tl = c(2, 2, 0,
2, 2, 0),
genotype = rep(c('WT', 'KO'), each = 3))
ezbado <- EZbakRArrowData(ds, metadf)Step 3: Run EstimateFractions like normal
Finally, just run EstimateFractions() with all of the
settings you would normally use if you were working with an
EZbakRData object:
ezbado <- EstimateFractions(ezbado,
features = c("GF", "XF",
"junction_start", "junction_end"),
filter_cols = c("XF", "junction_start",
"junction_end"),
filter_condition = `|`,
split_multi_features = TRUE,
multi_feature_cols = c("junction_start",
"junction_end"))Simulating the Arrow workflow
To give you the opportunity to explore using the Arrow backend in a
controlled setting, here is a workflow where we simulate a cB, create a
temporary arrow dataset with it, and run
EstimateFractions() on an EZbakRArrowData
object:
library(arrow)
#>
#> Attaching package: 'arrow'
#> The following object is masked from 'package:utils':
#>
#> timestamp
simdata <- EZSimulate(nfeatures = 250)
outdir <- tempdir()
dataset_dir <- file.path(outdir, "arrow_dataset")
write_dataset(
simdata$cB,
path = dataset_dir,
format = "parquet",
partitioning = "sample"
)
ds <- open_dataset(dataset_dir)
ezbdo <- EZbakRArrowData(ds,
simdata$metadf)
ezbdo <- EstimateFractions(ezbdo)
#> Estimating mutation rates
#> Estimating fractions for each sample:
#> ANALYZING sample1...
#> Extracting data for sample of interest and summarize out nucleotide content
#> Adding mutation rate estimation information
#> Estimating fractions
#> ANALYZING sample2...
#> Extracting data for sample of interest and summarize out nucleotide content
#> Adding mutation rate estimation information
#> Estimating fractions
#> ANALYZING sample3...
#> Extracting data for sample of interest and summarize out nucleotide content
#> Adding mutation rate estimation information
#> Estimating fractions
#> ANALYZING sample4...
#> Extracting data for sample of interest and summarize out nucleotide content
#> Adding mutation rate estimation information
#> Estimating fractions
#> ANALYZING sample5...
#> Extracting data for sample of interest and summarize out nucleotide content
#> Adding mutation rate estimation information
#> Estimating fractions
#> ANALYZING sample6...
#> Extracting data for sample of interest and summarize out nucleotide content
#> Adding mutation rate estimation information
#> Estimating fractions
#> ANALYZING sample7...
#> Counting reads
#>
#> ANALYZING sample8...
#> Counting reads
#>
#> Processing outputIn this case, a single sample is analyzed at a time to reduce RAM usage, as the arrow dataset allows EZbakR to only load the sample being currently analyzed into RAM. Note that this will result in a slight runtime hit relative to the non-arrow analysis. Thus, this is best reserved for working with large datasets.